- Home
- Genomes
- Genome Browser
- Tools
- Mirrors
- Downloads
- My Data
- Projects
- Help
- About Us
COVID-19 Research at UCSC Updated: Jan. 12, 2022
The SARS-CoV-2 coronavirus emerged in December 2019 as a novel human pathogen causing a severe acute respiratory syndrome (COVID-19). The disease spread rapidly worldwide and was declared a pandemic by the World Health Organization on March 11, 2020.
Genome sequencing of thousands of viral samples has helped researchers study mechanisms of infection, transmission and response of the human immune system. We at the UC Santa Cruz Genomics Institute are responding to the urgency of biomedical research to develop treaments and vaccines for this devastating illness by fast-tracking visualization of genome sequences and analyses in the UCSC Genome Browser for SARS-CoV-2. We are also incorporating relevant biomedical datasets such as single-cell lung gene expression into the UCSC Cell Browser, and creating data tracks of COVID-19 annotations in our Human Genome Browsers. These annotations can further understanding of the human genetic determinants of infection susceptibility, disease severity, and outcomes.
Since the beginning of SARS-CoV-2 circulation in humans, the viral genome has been accummulating mutations. Mutations identified as important medically and epidemiologically are displayed in SARS-CoV-2 browser tracks Variants of Concern and Spike Mutations. Investigations of antibody resistance of viral mutations are available in the Antibody Escape track collection. The alignment of the Pfizer and Moderna mRNA vaccine sequences to the SARS-CoV-2 genome can be viewed using the Vaccines track.
See our SARS-CoV-2 introduction page for an overview of the resources offered. A manuscript describing The UCSC SARS-CoV-2 Genome Browser was also published in the September 9, 2020 issue of Nature Genetics.

COVID-19 transmission as of May 30, 2020
Latest global analysis from Nextstrain.org
Latest global analysis from Nextstrain.org
UCSC Genome Browser view of SARS-CoV-2 genomic datasets

Video tutorial: Using the coronavirus browser.
COVID-19 and Lung gene expression data in the UCSC Cell Browser:
COVID-19 Datasets:
UShER for real-time genomic contact tracing
![]() In addition to the Genome Browser, we offer a
web interface to
Ultrafast Sample placement
on Existing tRees (UShER)
(Turakhia et al.),
a tool for identifying the relationships among a user's newly sequenced viral genomes
and all known SARS-CoV-2 virus genomes. UShER identifies relationships between viral
genomes by adding them to an existing phylogenetic tree of similar sequences that
visually depicts the evolutionary relationships among the genome sequences.
This approach empowers "genomic contact tracing".
That is, UShER tells you whether virus genomes are closely related and therefore
possibly from the same source, or if they are distantly related and the infections
come from distinct sources.
When newly sequenced virus genomes are added to a comprehensive tree of previously
sequenced genomes, contact tracers are often able to determine where in the world
those genomes came from.
UShER is the only available method that can do this in "real time".
Our tool places genomes onto a comprehensive global phylogeny of more than 80,000
virus genomes in less than one second.
More information about UShER can be found on the UCSC news article,
New tools enable rapid
analysis of coronavirus sequences and tracking of variants. The number of genome
sequences available has increased to 5.6 million since the article was initially published
with 1.2 million sequences on May 2021.
In addition to the Genome Browser, we offer a
web interface to
Ultrafast Sample placement
on Existing tRees (UShER)
(Turakhia et al.),
a tool for identifying the relationships among a user's newly sequenced viral genomes
and all known SARS-CoV-2 virus genomes. UShER identifies relationships between viral
genomes by adding them to an existing phylogenetic tree of similar sequences that
visually depicts the evolutionary relationships among the genome sequences.
This approach empowers "genomic contact tracing".
That is, UShER tells you whether virus genomes are closely related and therefore
possibly from the same source, or if they are distantly related and the infections
come from distinct sources.
When newly sequenced virus genomes are added to a comprehensive tree of previously
sequenced genomes, contact tracers are often able to determine where in the world
those genomes came from.
UShER is the only available method that can do this in "real time".
Our tool places genomes onto a comprehensive global phylogeny of more than 80,000
virus genomes in less than one second.
More information about UShER can be found on the UCSC news article,
New tools enable rapid
analysis of coronavirus sequences and tracking of variants. The number of genome
sequences available has increased to 5.6 million since the article was initially published
with 1.2 million sequences on May 2021.
Visit our YouTube channel for more videos.
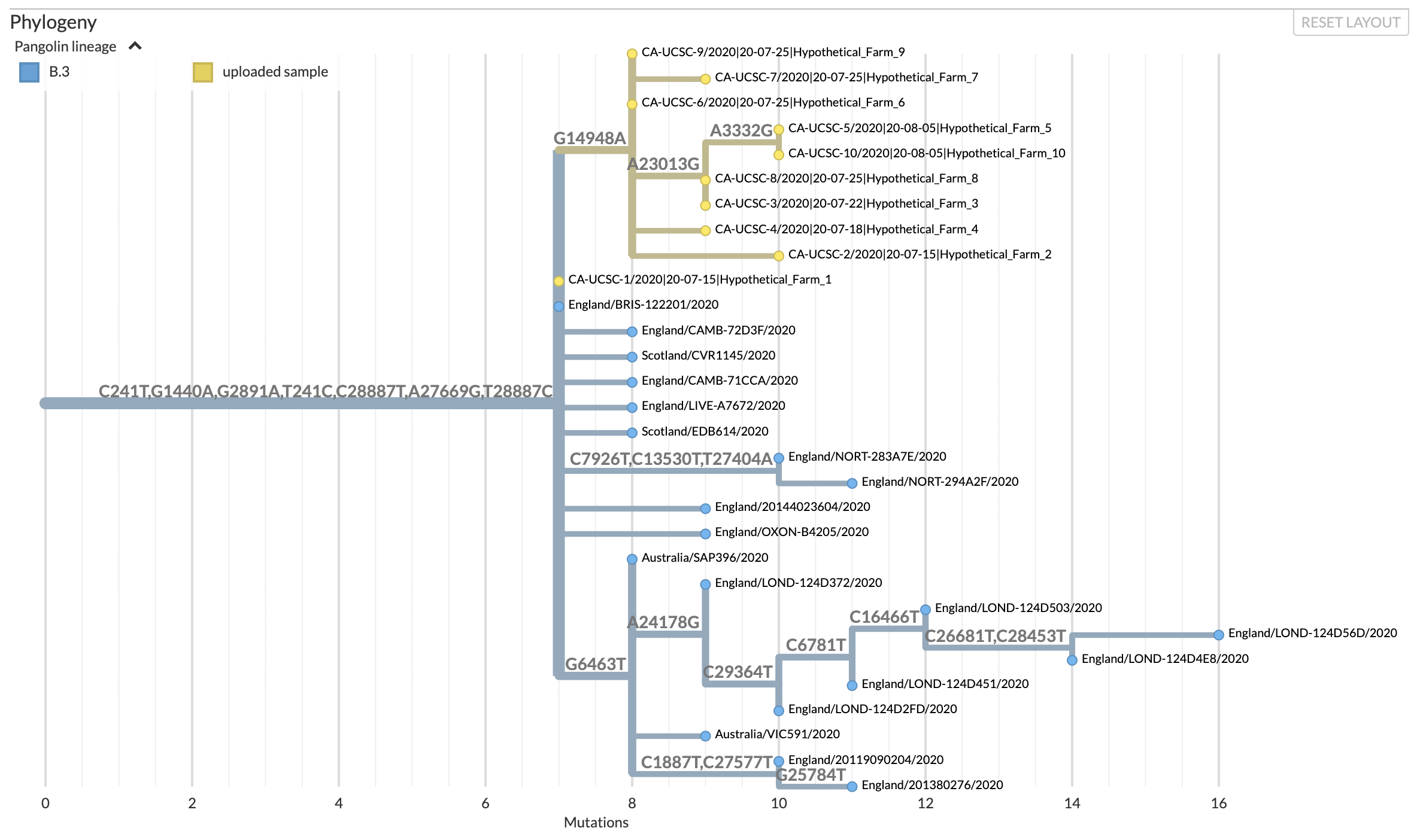
Example UShER results displayed using
Nextstrain.
Sequences representing a hypothetical outbreak are yellow;
previously sampled sequences are blue. Branches are labeled
by nucleotide mutations.
News
- January 12, 2022: Addition of the ARTIC V4.1 Oxford Nanopore sequencing primers. This track shows the primers for the ARTIC network SARS-CoV-2 sequencing protocol, Version 4.1 (January 7, 2022), which restores the funtionality of some primers against the Omicron variant.
- December 7, 2021: Updates to the SARS-CoV-2 Variants of Concern (VOC) track with amino acid and nucleotide annotations for 11 different COVID variants, including the Delta (B.1.617.2), Mu (B.1.621), and Omicron (B.1.1.529) variants.
- May 11, 2021: Release of SARS-CoV-2 Vaccines track, displaying the alignment of three different mRNA vaccine sequences to the coronavirus genome: the BioNTech/Pfizer BNT-162b2 sequence, the reconstructed BioNTech/Pfizer BNT-162b2 RNA sequence, and the Moderna mRNA-1273 sequence.
- February 17, 2021: Release of SARS-CoV-2 Variants of Concern track, displaying amino acid and nucelotide mutations in three "Variants of Concern" (B.1.1.7, B.1.351, and P.1) and one "Variant of Interest" (B.1.429, first identified in California), as defined in late January 2021.
- January 29, 2021: January 29th SARS-CoV-2 update - This update includes new data on sequence variants and phylogeny, spike protein amino-acid mutations, and antibody escape.
- January 8, 2021: January 8th SARS-CoV-2 update - This update includes an updated track as well as three new tracks for the coronavirus genome browser, and a track for the hg19/GRCh37 and hg38/GRCh38 human assemblies.
- October 21, 2020: New gene models, immunology, pathogenicity, and conservation annotation tracks have been released. Click the above link to directly access track description pages for specific track titles such as PhyloCSF Genes, Weizman ORFs, icSHAPE RNA Structure, Validated epitopes from IEDB, and various Phylogenetic Tree tracks.
- September 22, 2020: First COVID-19-related annotation track on UCSC human genome browser: COVID-19 GWAS: GWAS meta-analyses of studies contributed by multiple partners world-wide (18 in this release), as part of the COVID-19 Host Genetics Initiative, a collaborative effort to facilitate the generation, analysis and sharing of COVID-19 host genetics research.
- September 9, 2020: The manuscript describing the The UCSC SARS-CoV-2 Genome Browser, is now published in Nature Genetics.
- August 7, 2020: Eight new annotation tracks, presenting information about duplication regions, sequencing, gene identities, plasmid constructs, protein contact sites, and variant distribution.
- July 7, 2020: Annotation tracks released: PhyloCSF (Codon Substitution Frequency), Microdeletions, and Human CoV (multiple alignment of SARS-CoV-2, SARS-CoV-1, MERS, and three other human-infecting coronaviruses)
- May 29, 2020: Public track hub GENCODE annotation of COVID-19 associated genes released. View in browser.
- Video: Coronavirus Browser SARS CoV-2
- May data release for SARS-CoV-2 genome browser
- April data release for SARS-CoV-2 genome browser
- February 7, 2020: Released initial SARS-CoV-2 genome browser
Related publications and data resources
- Preprints of research manuscripts: BioRxiv/MedRxiv COVID-19 Collection (all freely available)
- COVID-19 Open Research Dataset of scholarly literature
- GISAID the main repository of SARS-CoV-2 sequences
- NIH Office of Data Science Strategy Open-Access Data and Computational Resources to Address COVID-19
- European Molecular Biology Laboratory/European Bioinformatics Institute (EMBL-EBI) and partners: COVID-19 Data Portal
- China National Center for Bioinformation: 2019 Novel Coronavirus Resource
- CDC SARS-CoV-2 Sequencing Resources on GitHub
- Nextstrain open-source tracking of pathogen evolution: SARS-CoV-2 browser (strain tree, geographical mapping)
- NCBI sequence database search, publication list, and genome display: NCBI SARS-CoV-2 Sequences
- Virus Pathogen Resource: VIPR SARS-CoV-2 resource (tools for sequence and structure analysis, comparative genomics and phenotype studies)
- CDC: Cases in U.S., updated daily.
- Johns Hopkins University: Coronavirus Resource Center
- CitizenScience.org: COVID-19 citizen science and crowdsourcing projects
- Wikipedia: Severe acute respiratory syndrome coronavirus 2
- COVID Textbook: COVID Reference
- Nature News graphical guide to vaccine designs: The Race for Coronavirus Vaccines
- Inside the Coronavirus Genome, and How Coronavirus Mutates and Spreads, The New York Times
- UCSC's Million-COVID-Genome Tree Could be a First, UCSC Genomics Institute
- SARS-CoV-2 gene content and COVID-19 mutation impact by comparing 44 Sarbecovirus genomes, Nature Genetics.
Funding
The UCSC Genome Browser is a publicly available web resource serving the research community since July 2000. Both the Genome Browser and UCSC Cell Browser are funded by the NIH National Human Genome Research Institute. The Genome Browser recieved funding from the NIAID, National Institute of Allergy and Infectious Disease. In addition to this funding, the COVID-19 projects here are funded by generous supporters including: several anonymous donors; Pat and Roland Rebele; Eric and Wendy Schmidt by recommendation of the Schmidt Futures program; the Center for Information Technology Research in the Interest of Society (CITRIS); and the University of California Office of the President (UCOP). To contribute, click here.