Description: Homo sapiens DEAD (Asp-Glu-Ala-Asp) box helicase 1 (DDX1), mRNA. RefSeq Summary (NM_004939): DEAD box proteins, characterized by the conserved motif Asp-Glu-Ala-Asp (DEAD), are putative RNA helicases. They are implicated in a number of cellular processes involving alteration of RNA secondary structure such as translation initiation, nuclear and mitochondrial splicing, and ribosome and spliceosome assembly. Based on their distribution patterns, some members of this family are believed to be involved in embryogenesis, spermatogenesis, and cellular growth and division. This gene encodes a DEAD box protein of unknown function. It shows high transcription levels in 2 retinoblastoma cell lines and in tissues of neuroectodermal origin. [provided by RefSeq, Jul 2008]. Transcript (Including UTRs) Position: hg19 chr2:15,731,745-15,771,235 Size: 39,491 Total Exon Count: 26 Strand: + Coding Region Position: hg19 chr2:15,732,058-15,771,030 Size: 38,973 Coding Exon Count: 26
ID:DDX1_HUMAN DESCRIPTION: RecName: Full=ATP-dependent RNA helicase DDX1; EC=3.6.4.13; AltName: Full=DEAD box protein 1; AltName: Full=DEAD box protein retinoblastoma; Short=DBP-RB; FUNCTION: Acts as an ATP-dependent RNA helicase, able to unwind both RNA-RNA and RNA-DNA duplexes. Possesses 5' single-stranded RNA overhang nuclease activity. Possesses ATPase activity on various RNA, but not DNA polynucleotides. May play a role in RNA clearance at DNA double-strand breaks (DSBs), thereby facilitating the template-guided repair of transcriptionally active regions of the genome. Together with RELA, acts as a coactivator to enhance NF-kappa-B-mediated transcriptional activation. Acts as a positive transcriptional regulator of cyclin CCND2 expression. Binds to the cyclin CCND2 promoter region. Associates with chromatin at the NF- kappa-B promoter region via association with RELA. Binds to poly(A) RNA. May be involved in 3'-end cleavage and polyadenylation of pre-mRNAs. Required for HIV-1 Rev function as well as for HIV-1 replication. Binds to the RRE sequence of HIV-1 mRNAs. CATALYTIC ACTIVITY: ATP + H(2)O = ADP + phosphate. SUBUNIT: Interacts with PHF5A (via C-terminus) (By similarity). Interacts with MBNL1. Interacts with CSTF2. Interacts with HNRNPK. Interacts with ATM. Interacts with RELA (via C-terminus). Interacts (via C-terminus) with the replicase polyprotein 1ab Nsp14 of the avian infectious bronchitis virus (IBV). Interacts with Rev of HIV-1. Interacts with severe acute respiratory syndrome coronavirus (SARS-CoV) (via N-terminus). Component of the tRNA-splicing ligase complex. SUBCELLULAR LOCATION: Nucleus. Cytoplasm. Cytoplasmic granule. Note=Localized with MBNL1, TIAL1 and YBX1 in stress granules upon stress. Localized with CSTF2 in cleavage bodies. Forms large aggregates called DDX1 bodies. Relocalized into multiple foci (IR- induced foci or IRIF) after IR treatment, a process that depends on the presence of chromosomal DNA and/or RNA-DNA duplexes. Relocalized at sites of DNA double-strand breaks (DSBs) in an ATM- dependent manner after IR treatment. Colocalized with RELA in the nucleus upon TNF-alpha induction. Relocalized to the cytoplasm with a perinuclear staining pattern in avian infectious bronchitis virus (IBV)-infected cells. Required for proper localization of HIV-1 Rev. TISSUE SPECIFICITY: Highest levels of transcription in 2 retinoblastoma cell lines and in tissues of neuroectodermal origin including the retina, brain, and spinal cord. DOMAIN: The helicase domain is involved in the stimulation of RELA transcriptional activity. PTM: Phosphorylated. Phosphorylated by ATM kinase; phosphorylation is increased in response to ionizing radiation (IR). SIMILARITY: Belongs to the DEAD box helicase family. DDX1 subfamily. SIMILARITY: Contains 1 B30.2/SPRY domain. SIMILARITY: Contains 1 helicase ATP-binding domain. SIMILARITY: Contains 1 helicase C-terminal domain. CAUTION: According to some authors the unwinding activity is ADP- dependent and not ATP-dependent (PubMed:18710941).
Cholesterol, LDL Sekar Kathiresan et al. BMC medical genetics 2007, A genome-wide association study for blood lipid phenotypes in the Framingham Heart Study., BMC medical genetics.
[PubMed 17903299]
Using a 100K genome-wide scan, we have generated a set of putative associations for common sequence variants and lipid phenotypes. Validation of selected hypotheses in additional samples did not identify any new loci underlying variability in blood lipids. Lack of replication may be due to inadequate statistical power to detect modest quantitative trait locus effects (i.e., <1% of trait variance explained) or reduced genomic coverage of the 100K array. GWAS in FHS using a denser genome-wide genotyping platform and a better-powered replication strategy may identify novel loci underlying blood lipids.
Hair Sarah E Medland et al. American journal of human genetics 2009, Common variants in the trichohyalin gene are associated with straight hair in Europeans., American journal of human genetics.
[PubMed 19896111]
Hip Douglas P Kiel et al. BMC medical genetics 2007, Genome-wide association with bone mass and geometry in the Framingham Heart Study., BMC medical genetics.
[PubMed 17903296]
The FHS 100K SNP project offers an unbiased genome-wide strategy to identify new candidate loci and to replicate previously suggested candidate genes for osteoporosis.
The RNAfold program from the Vienna RNA Package is used to perform the secondary structure predictions and folding calculations. The estimated folding energy is in kcal/mol. The more negative the energy, the more secondary structure the RNA is likely to have.
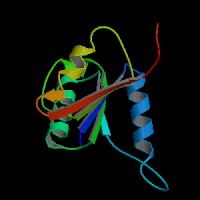
ModBase Predicted Comparative 3D Structure on Q92499
Front
Top
Side
The pictures above may be empty if there is no ModBase structure for the protein. The ModBase structure frequently covers just a fragment of the protein. You may be asked to log onto ModBase the first time you click on the pictures. It is simplest after logging in to just click on the picture again to get to the specific info on that model.
Orthologous Genes in Other Species
Orthologies between human, mouse, and rat are computed by taking the best BLASTP hit, and filtering out non-syntenic hits. For more distant species reciprocal-best BLASTP hits are used. Note that the absence of an ortholog in the table below may reflect incomplete annotations in the other species rather than a true absence of the orthologous gene.